[gtranslate]
Antiperovskitas: Estrutura e Eletrônica por DFT | |
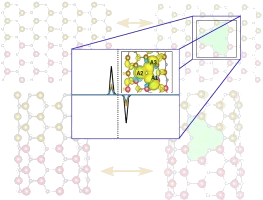
Focada em antiperovskitas (óxidos, nitretos e haletos), esta linha busca elucidar relações entre estrutura cristalina, estabilidade e propriedades de transporte iônico/eletrônico visando aplicações em eletrólitos sólidos, sensores e conversão de energia. A investigação inclui cálculo de superfícies de energia potencial para migração iônica (vacâncias/intersticiais), elasticidade e resposta mecânica, propriedades eletrônicas (band structure/PDOS) e estabilidade vibracional por fônons, além do efeito de dopagem, pressão e temperatura. Os resultados orientam a seleção de composições com baixas barreiras de migração, janelas eletroquímicas adequadas e robustez mecânica. Dentre as ferramentas computacionais utilizadas destacam-se: VASP e Quantum ESPRESSO (DFT), PHONOPY (fônons). |
Publicações relacionadas:
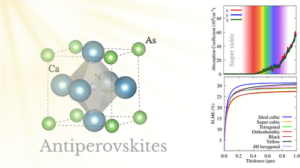
Seletividade e Transporte de Gases em Zeólitas por MD e DFT | |
Esta linha combina DFT e MD para estudar adsorção e transporte seletivo de moléculas (p.ex., CO₂/CH₄/N₂/H₂O) em redes microporosas de zeólitas, com ênfase em estruturas do grupo CHA (e correlatas). O trabalho aborda localização de sítios ativos, efeito da distribuição de heteroátomos/cátions, competição multicomponente, difusão em canais estreitos e estabilidade em presença de umidade. São estimadas energias de adsorção, perfis de energia livre e coeficientes de difusão que fundamentam a previsão de seletividade e permeabilidade, orientando o desenho de processos de separação e captura com menor custo energético. Dentre as ferramentas computacionais utilizadas destacam-se: SIESTA/VASP (DFT), LAMMPS (MD) e rotinas de análise em Python; quando aplicável, simulações de GCMC complementam a obtenção de isotermas. |
Publicações relacionadas:

Materiais de Eletrodo para Baterias de Íons de Lítio por DFT | |
O objetivo desta linha é avaliar, por DFT, novos materiais de ânodo e cátodo, elucidar rotas de inserção/extração de Li e antecipar métricas de desempenho relevantes para baterias de alta densidade energética. São investigados energias de adsorção e intercalação, tensões de circuito aberto, barreiras de migração iônica (via NEB), estabilidade estrutural (inclusive sob variação volumétrica) e integridade mecânica, além do papel de defeitos e dopagem na condutividade eletrônica/ iônica. A abordagem prioriza correlação entre estrutura atômica, termodinâmica e transporte, subsidiando o screening de composições com maior capacidade específica e segurança operacional. Dentre as ferramentas computacionais utilizadas destacam-se: VASP e SIESTA (DFT), PHONOPY (estabilidade vibracional), NEB/CI-NEB (difusão) e VASPKIT/ASE (análise). |

Dinâmica Molecular de Supercapacitores e Interfaces Eletroquímicas | |
Esta linha se destina a compreender, por Dinâmica Molecular clássica, os mecanismos de armazenamento eletrostático em supercapacitores, com foco na formação da camada dupla elétrica em interfaces carbono–eletrólito. O trabalho investiga o impacto da textura do eletrodo (microporosidade/mesoporosidade), da composição do eletrólito (aquoso, orgânico, water-in-salt, líquidos iônicos) e da temperatura sobre estrutura iônica, difusividade, cinética de adsorção e resposta dinâmica. Metodologias de carga constante e potencial constante são empregadas para aproximar condições reais de operação e extrair métricas como capacitância diferencial, resistência de transferência e estabilidade de ciclagem — insumos diretos para otimização de desempenho e vida útil do dispositivo. Dentre as ferramentas computacionais utilizadas destacam-se: LAMMPS (MD), PACKMOL (preparação estrutural) e OVITO/Python (análise). |

Modelagem Ab Initio de MXenes para Conversão de Energia | |
Esta linha investiga MXenes bidimensionais (e suas funcionalizações superficiais) como plataformas para conversão de energia e catálise eletroquímica, visando processos como redução de CO₂, evolução/oxidação de H₂ e reações redox em dispositivos. O estudo abrange estabilidade termodinâmica e dinâmica, propriedades eletrônicas, função trabalho, sítios ativos e o efeito de defeitos, dopagem e tensão mecânica na energia de adsorção de intermediários reacionais. A ênfase recai na identificação de descritores (por exemplo, ΔG de adsorção) que correlacionem estrutura e desempenho, possibilitando o desenho racional de materiais mais eficientes e duráveis em condições operacionais. Dentre as ferramentas computacionais utilizadas destacam-se: VASP, SIESTA e Quantum ESPRESSO (DFT), com apoio de PHONOPY (fônons) e VASPKIT/ASE (pós-processamento). |

Modelagem e Simulação de Processos Optoeletrônicos em Semicondutores Orgânicos | |
Por meio de métodos de química quântica e física computacional, este projeto se propõe à modelagem computacional de diversos sistemas orgânicos a serem utilizados como camadas ativas em dispositivos optoeletrônicos, especialmente em sistemas fotovoltaicos. A investigação de processos de transferência, separação e recombinação de portadores de carga em heterojunções orgânicas, sistemas que podem ser usados no desenvolvimento de células solares (dispositivos fotovoltaicos), será o principal objeto de estudo. Considerável parte deste projeto é dedicada ao desenvolvimento de novas metodologias capazes de tornar as condições de simulação mais realistas e os resultados mais acurados, abrangendo, portanto, um viés metodológico. O desenvolvimento dessas novas metodologias pode proporcionar uma descrição mais detalhada e precisa da estrutura eletrônica de vários sistemas orgânicos, tanto em nível atômico quanto na escala molecular, contribuindo para um melhor entendimento dos processos físicos envolvidos no funcionamento dos dispositivos optoeletrônicos e, também, para o desenvolvimento de novos materiais, o que é muito atrativo dos pontos de vista acadêmico e industrial. Cálculos de dinâmica molecular serão empregados para estudar heterojunções orgânicas em candidatos a sistemas fotovoltaicos de alto desempenho. Combinando os cálculos de dinâmica molecular e mecânica quântica, será possível propor uma descrição mais realista dos problemas de transporte, transferência, separação e recombinação de carga em várias classes de condutores orgânicos. Também, entre os problemas estudados, destaca-se a dinâmica de estados excitados, como o transporte de polarons, por exemplo, em polímeros conjugados e em nanofitas de grafeno. O problema da dinâmica de estados excitados em sistemas orgânicos é investigado no escopo de modelos Tight-Binding (em português Ligação Rígida) com relaxação em uma e duas dimensões. |
|
Uso da Dinâmica Molecular Reativa para o Estudo das Propriedades Físico-Químicas de Novas Nanoestruturas |
|
|
Esta linha se destina ao estudo das propriedades físico-químicas de nanoestruturas baseadas em novos alótropos de carbono, buscando propor materiais mais eficientes no que tange às aplicações de conversão e armazenamento de energia. Entre os problemas estudados destacam-se as propriedades estruturais, mecânicas, padrões de fraturas e degradação em atmosferas gasosas destes sistemas nas formas de monocamada, tubo e scroll. Considerável parte desse projeto é dedicada ao desenvolvimento de novas metodologias, baseadas em dinâmica molecular reativa, capazes de tornar as condições de simulação mais realistas e os resultados mais acurados para uma boa descrição dos dados fornecidos pelos experimentos na literatura. A dinâmica molecular, com o uso de um potencial reativo, permite estudar a dissociação e a formação de ligações químicas em sistemas nanoestruturados. O desenvolvimento dessas novas metodologias baseadas em dinâmica molecular pode proporcionar elementos para uma descrição mais detalhada e precisa da estrutura eletrônica desses sistemas tanto em nível atômico quanto na escala molecular, contribuindo para um melhor entendimento dos processos físico-químicos envolvidos no funcionamento de dispositivos optoeletrônicos, também, para o desenvolvimento de novos materiais, o que é muito atrativo dos pontos de vista acadêmico e industrial. Dentre as ferramentas computacionais utilizadas nessa linha de pesquisa destacam-se: LAMMPS (potenciais ReaxFF, AIREBO, Tersoff e Stillinger-Weber (SW)) e Materials Studio (Módulo GULP). |
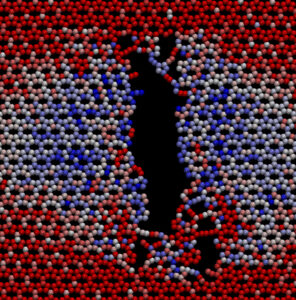
Uso da Teorial do Funcional de Densidade para o Estudo da Estrutura Eletrônica de Nanomateriais | |
Nesta linha de pesquisa, estudamos a estrutura eletrônica de nanomateriais que têm sido empregados no desenvolvimento de dispositivos optoeletrônicos. A investigação das propriedades ópticas, estruturais e eletrônicas de monocamadas e bicamadas (também na forma de heteroestruturas) compostas de dicalcogenetos de metal de transição (TMDS, do inglês Transition Metal Dichalcogenides), grafeno e seus alótropos, perovskitas e nitretos do grupo III é o principal foco do projeto. Considerável parte deste projeto é dedicada ao design de novos materiais – buscando uma descrição realista e detalhada da estrutura eletrônica desses novos sistemas – que promovam um aumento na eficiência de funcionamento de dispositivos optoeletrônicos de grande interesse acadêmico e industrial, tais como transistores de filme fino, diodos emissores luz e células fotovoltaicas. Dentre as ferramentas computacionais utilizadas nessa linha de pesquisa destacam-se: SIESTA, QUAMTUM ESPRESSO e Materials Studio (Módulos CASTEP e DMol3). |
Publicações relacionadas:
Physica E: Low-dimensinal Systems ans Nanostrucutres, 130, 114683, 2021
Chemical Physics Letters, 771, 138495, 2021
Electronic Structure, 3, 024005, 2021
Physical Chemistry Chemical Physics, 23, 18807-10813, 2021
Computational Materials Science, 183, 109860, 2020
Physical Chemistry Chemical Physics, 21, 11168-11174, 2019